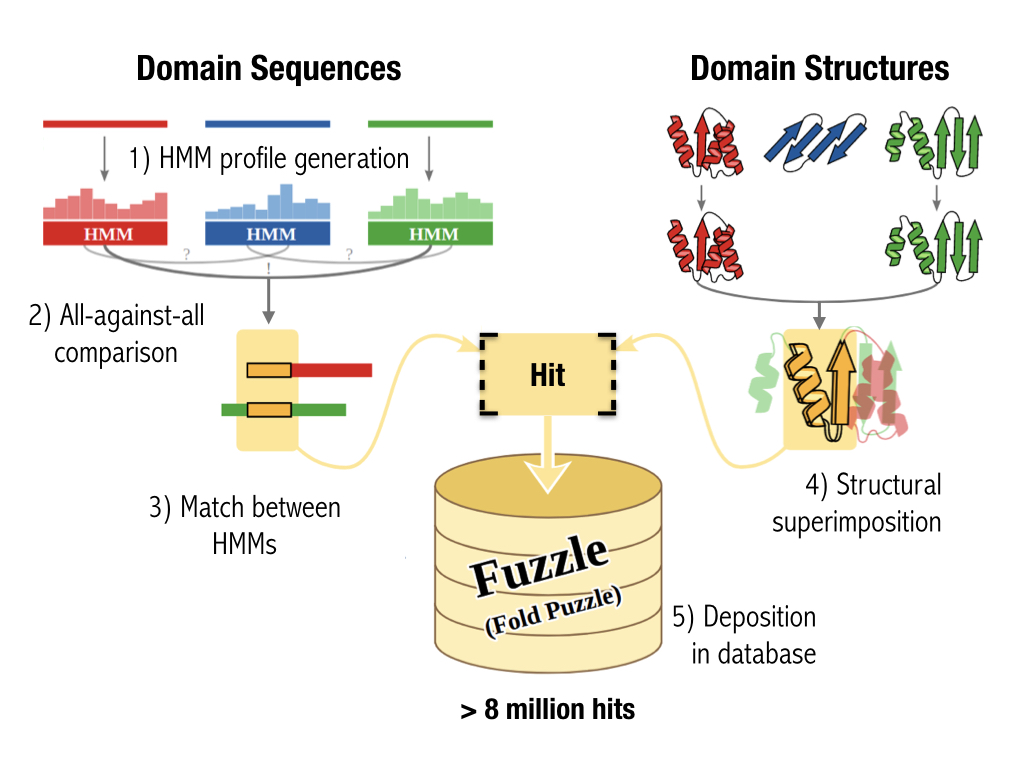
To build a database among different domains
we first needed an input database of protein structures classified into folds.
We used the SCOPe database, releases 2.03 to 2.07.
The SCOPe database is a database of domains of known structure classified according
to structural and evolutionary relationships.
It classifies protein domains hierarchically into levels, the two lowest (family and superfamily) capturing
homologous relationships, while the upper levels, folds and classes, group superfamilies according to their topological connections.
Let's specify the process we did for SCOPe 2.06. We took the SCOP95 subset, where sequences are filtered out to keep a maximum of 95% identity between any two domains.
This input database contains 28010 domains. For each domain sequence we created a sequence alignment either using
PSIblast.
The multiple alignments served as base to build a profile HMM for each domain.
We then performed an all-against-all pairwise HMM profile
comparisons of the full domains in the sequence space alone, using HHsearch.
At this stage, we had pairs of domains that share a protein segment with a certain homology.
Subsequently, we measured the structural similarity of each pair using
TM-align.
We refer to these protein segments as fragments , i.e,
significantly sized protein segments with remote homology and
similar structure. This pipeline led to a final set of over 8 million pairs (termed hits herein)
that we stored into our Fuzzle database, standing for Fold Puzzle..
Pipeline to construct the database.
Input domain sequences and structures were taken from the SCOPe database.
Profile-HMMs were built after generating a multiple sequence alignment for each sequence.
The profile-HMM were all-against-all compared for sequence homology.
Matches between pairs of domains were also structurally superimposed.
These structural and sequece alignment constitutes one Fuzzle hit.
The database contains 8 millions.
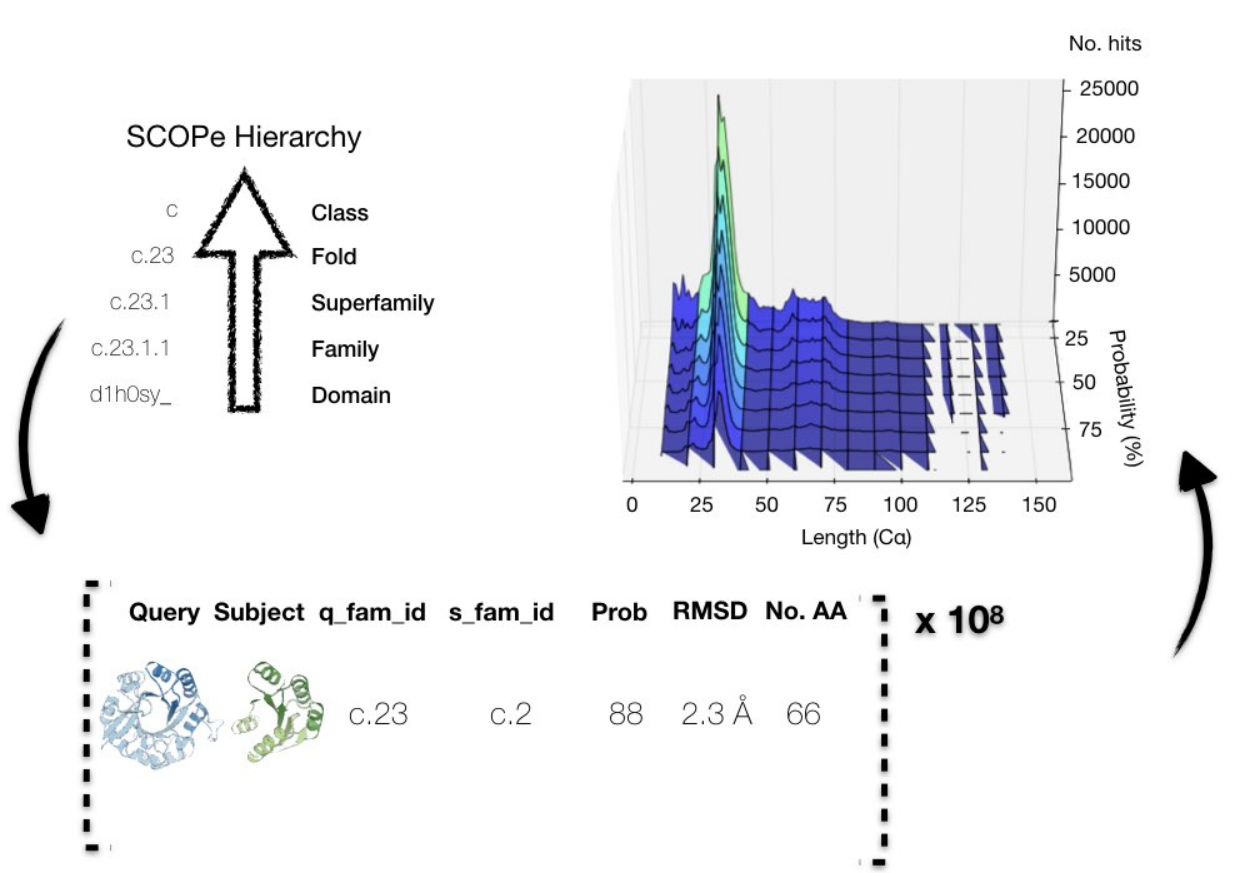
A look inside the database
The SCOPe database served as input to construct Fuzzle.
Each hit on fuzzle contains information about the two domains
(query and subject), HHsearch probability, and RMSD, among other parameters.
The more than 8 million hits define a space of protein sequence and structural similarities
that can be visualized in many ways, such as by projecting them by their length and probability.
Most of the fragments are short (lengths < 50) and present low HHsearch probabilities (<50%),
but the amount of other hits is nevertheless non-negligible.
Internally, each hit is comprised by the two domain SCOP identifiers
(query and subject), their SCOPe families, the HHsearch probability of the homologous protein
fragment, sequence positions of the fragment, and the TM-score and RMSD of the structural alignment
for the sequence fragment (see figure above).
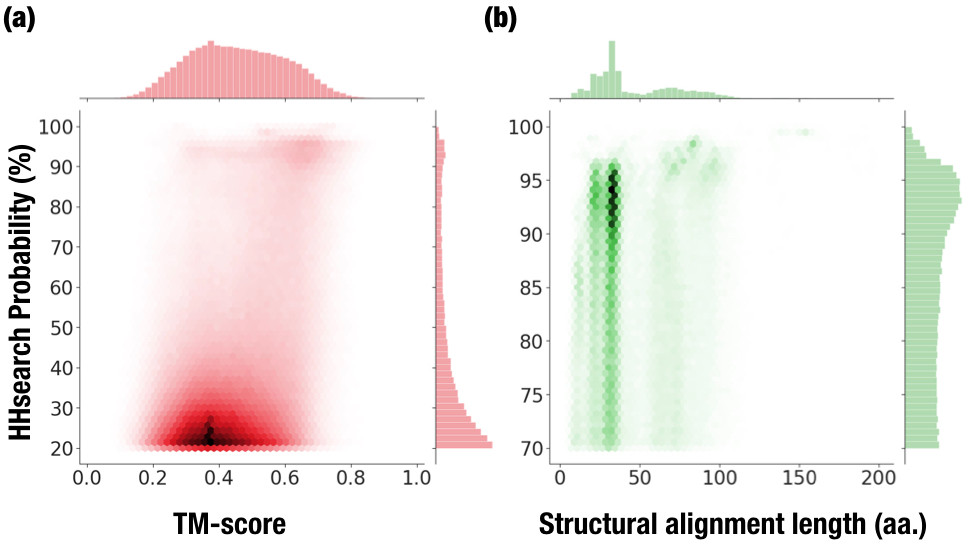
There are over 1.8 million hits where the two domains belong to different folds.
We observed a bimodal distribution when plotting TM-score vs. HHsearch probabilities for pairs with domains belonging to different folds
(high probability, high TM-score):
Statistics of Fuzzle based on SCOPe 2.06. (a) HHsearch probabilities vs. TM-score
for Fuzzle hits belonging to different folds. (b) HHsearch probability vs. the length of the structural alignment for hits
where TM-score > 0.3 and HHsearch probability > 70%
The set of hits in the upper left of the figure, i.e hits with high-sequence and structural similarity, reveals that
that there are unrecognized homologous relationships.
We therefore explored further this set of hits, those with probabilities over 70% and TM-score > 0.3.
When representing this set of hits according to their length and probability (b),
we observe most of the hits present short lengths (0 - 40 amino acids) and low probabilities.
There are however, a non-negligible proportion of hits at larger lengths (50 – 70 amino acids)
and some small areas with fragments over 100 amino acids.